- Reviewed last night’s simulations, see yesterday’s post.
Simulated data testing:
Fitting the general model well on the parrotfish tree even with clear simulated data seems hard. Simulating with alpha(intramandibulars) = .01, and others = 20, and fitting general model and the alpha-only independent model (release of constraint model). The release model MLE recovers reasonable values: alpha(intra) = 1.7, alpha(others) = 21.8, while the general model fit is clearly struggling even with SANN.
[table id=3 /]
May help to try seeding with optima of the sub-models. This is expressed in the bootstraps of course, where the release model produces rather clear distributions (all with 80 reps):
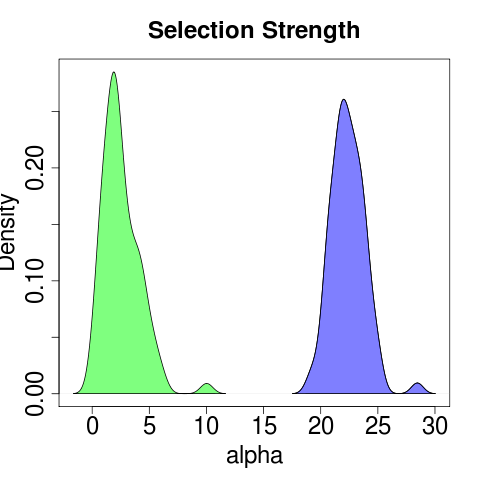
though the general model is much more confused:

Repeating with more replicates, but should try the remaining submodels. This obviously shows up in the model comparison by bootstrapped likelihood ratio:
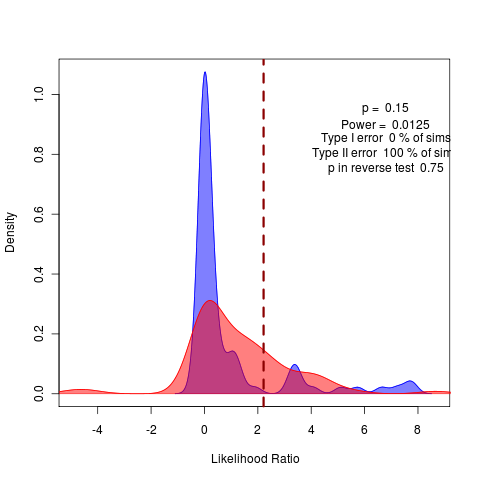
Note how crucial it is we did the test distribution. Without it, we’d think this a rather clear rejection of the variable alphas model in place of the general model.
MCMCs
MCMCs on these models. Having difficulty with undefined Hastings ratios, probably on out-of-bound proposals, though not sure why those aren’t being caught. Good to know the MLE runs with the same likelihood function.
MCMC errors are occurring in MCMCMC, dropping the heated chains function for the moment works fine. Some difficulty in recombining chain intervals in the coupling, will need a close look. FIXME.
Running long MCMCs on alpha (on zero, n8) and general (on one, n15) models. Combines 16 chains (minus burnin), of 1e5 reps. Simulated_mcmc.R example and sim_general_mcmc.R examples.
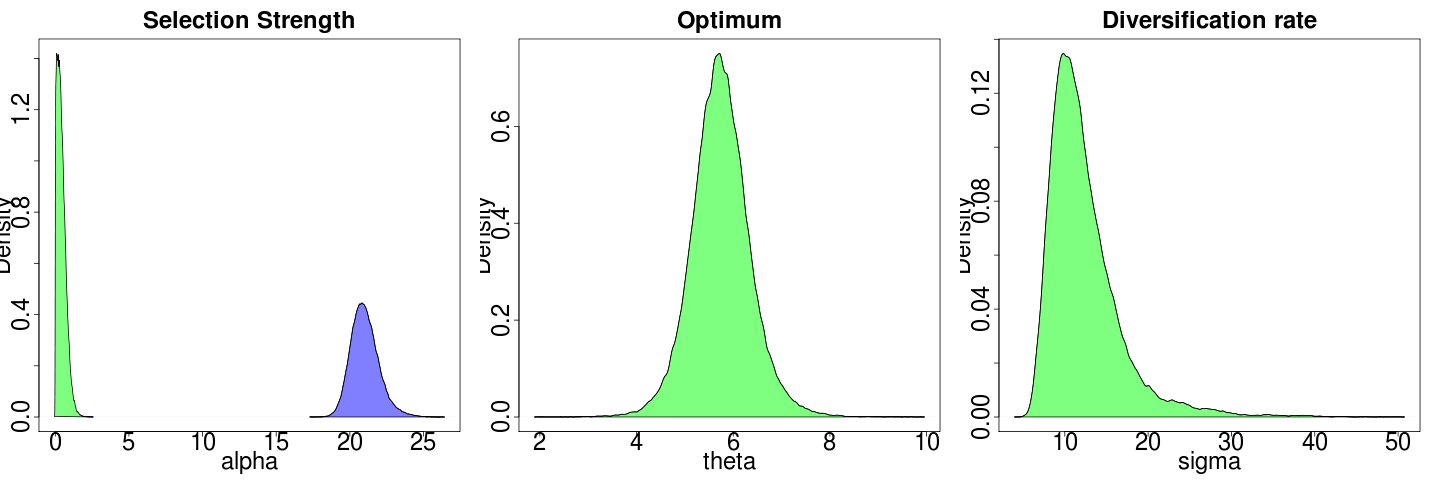
Ooh, much faster when not merging multiple chains. let’s run 16 x 1e6.
Max Likelihood + bootstraps
- Piecewise initialized simulated annealing from the submodels: Works! Also note: the aggressive SANN is still needed to get convergence:
> gen_fit <- smart_multiType(data=sim_trait, tree=labrid$tree,
+ regimes=intramandibular,Xo=NULL, alpha = .1,
+ sigma = .1, theta=NULL)
[1] "alpha: 3.02538925492894e-07" "alpha: 0.0585819705289946"
[1] "sigma: 0.00166254126803767" "sigma: 2.90960629757019"
[1] "theta: 1.23973641541187" "theta: 1.84059077604827"
>
> print(getParameters.multiOU(gen_fit))
alpha1 alpha2 theta1 theta2 sigma1 sigma2
0.756620314 3.758072410 13.732765801 4.853751270 0.006160961 3.913957909
Xo
4.667250491
>
>
> sann_fit <- smart_multiType(data=sim_trait, tree=labrid$tree,
+ regimes=intramandibular,Xo=NULL, alpha = .1,
+ sigma = .1, theta=NULL,
+ method ="SANN", control=list(maxit=80000,temp=25,tmax=50))
[1] "alpha: 0.271749815040882" "alpha: 20.4096202543866"
[1] "sigma: 0.0037342888296498" "sigma: 5.5349835595542"
[1] "theta: 4.81905476727201" "theta: 4.58842972148977"
>
> print(getParameters.multiOU(sann_fit))
alpha1 alpha2 theta1 theta2 sigma1 sigma2 Xo
0.2609878 21.9790836 -5.5837586 4.6213975 17.9955025 9.6907227 7.3360959
>
Should probably compare likelihoods of all models as well to check convergence.
Hmm, likelihood of sann_fit isn’t actually better than the very far off combo of nelder-mead? Change SANN starting temp?
Seems to be lack of power. Seeding from a theta-only estimate doesn’t help at all in this case, as chooses wildly different theta. meanwhile, sigma model is doing slightly better anyway:
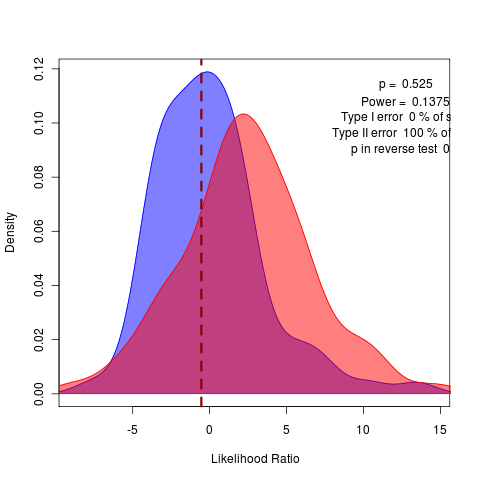
Each model fit creates reasonably tight bootstraps around its MLE estimates:

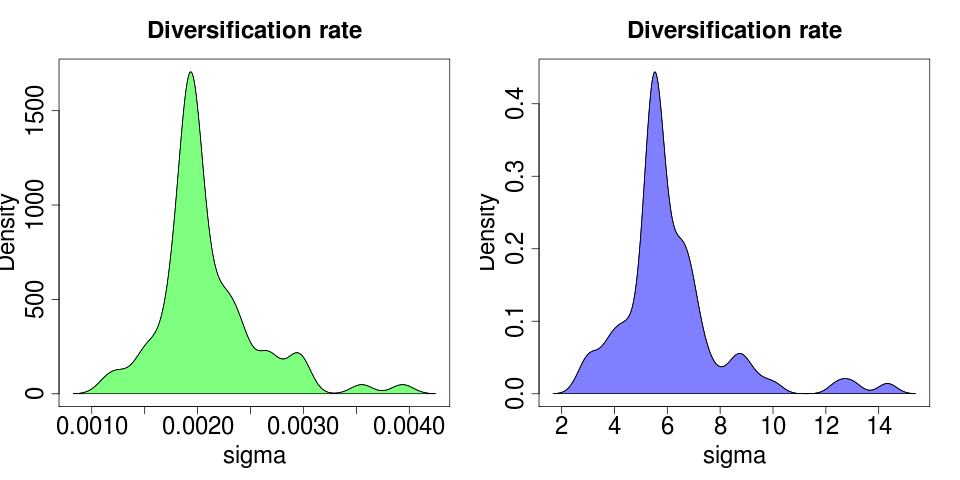
(plotted on different axes due the large difference in y max).
Fixing “fixed” parameter style/ brownie. has no alpha value stored, so can’t simulate. FIXED.
Try on larger (labrid? simulated?) tree, will need a reasonable painting defined as well. Seems to be a convergence problem, but more power may help tame the ridges.
Database projects
TreeBASE
iEvoBio talk accepted!
Johan Nylander’s advice: try readNexus() from phylobase package. Unfortunately no go so far. Can’t handle urls so download the file (needs wget interface). See today’s update onissue tracker.
RPLoS
Wrote an R function implementing CrossRef API for use in rplos.
Oooh, PLoS and Mendeley have combined forces in the competition to use their APIs. Wonder if they’ll be very responsive to feature requests on the API itself…